Introduction
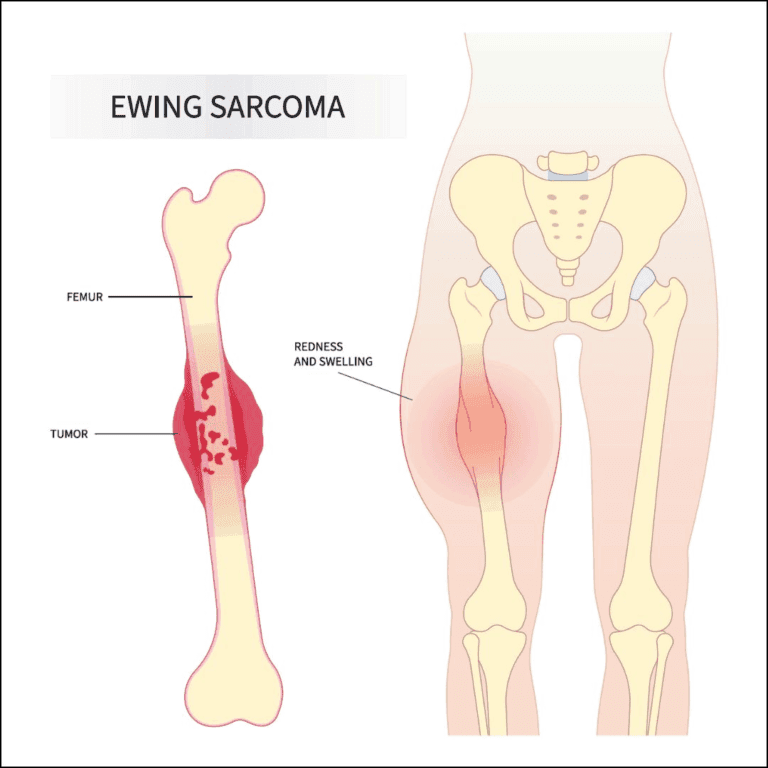
Ewing’s sarcoma is a malignant tumour that primarily starts in bones, often affecting children and young adults, particularly during their teenage years. While it can develop in any bone, it usually targets long bones like the femur, tibia, and humerus, as well as the pelvis. Occasionally, it originates in muscles and soft tissues, known as extraosseous Ewing’s sarcoma. This cancer can metastasize to the lungs, bone marrow, and other soft tissues.
Overview
Ewing’s sarcoma is rare compared to other cancers but is the second most common malignant bone tumour in children and young adults. About 1.7 out of 1 million children under 15 develop this disease. It is part of the Ewing’s Family of Tumours (EFT), which includes:
- Ewing’s sarcoma of bone
- Ewing’s sarcoma of soft tissue
- Primitive neuroectodermal tumour (PNET)
- Askin’s tumour (a PNET in the chest bones)
This article focuses on Ewing’s sarcoma of the bone.
Cause
The exact cause of Ewing’s sarcoma is unknown. Unlike most cancers that arise from specific tissues, the origin of Ewing’s sarcoma cells remains unclear. It involves a genetic abnormality where chromosomes #11 and #22 are mismatched. This mutation is not inherited but occurs after birth, with no identified risk factors or prevention methods.
Symptoms
Common symptoms include pain and swelling at the tumour site, which might not appear until the tumour grows significantly. Sometimes, a mass may be the first noticeable symptom. Injuries might highlight the presence of a tumour when weakened bones break easily.
Diagnosis
Diagnosing Ewing’s sarcoma involves:
- Imaging Tests: X-rays, MRI scans, CT scans, and bone scans to visualise the affected area.
- Biopsy: Confirming the tumour through tissue examination under a microscope. Ewing’s sarcoma cells are identified by their small, round, blue appearance.
Staging
Staging determines the cancer’s spread using blood tests, CT scans of the lungs, bone scans, and bone marrow biopsies. Understanding the primary and metastatic sites helps devise an effective treatment plan.
Treatment
Treatment typically involves a combination of chemotherapy, surgery, and radiation. Specialists from various fields collaborate to tailor treatments to individual cases.
Chemotherapy:
Used to shrink the primary tumour and treat potential spread. It involves cycles of drug combinations over about a year, with side effects managed by specific medications.
Surgery:
Removes the primary tumour and reconstructs affected bones and tissues using grafts and artificial joints. Physical therapy aids recovery and functionality.
Radiation:
Destroys cancer cells, especially when surgery isn’t feasible. However, it carries risks like skin damage, muscle scarring, and potential secondary cancers.
Outcomes
Advancements in chemotherapy, imaging, and surgical techniques have improved survival rates. Approximately two-thirds of patients without metastasis survive at least five years post-diagnosis. Continuous follow-up is crucial to monitor for late side effects and recurrence.
Research
Ongoing research aims to understand the genetic abnormalities in Ewing’s sarcoma, improve chemotherapy protocols, and enhance surgical and prosthetic techniques. Innovations like expandable prostheses and tissue growth factors are promising future treatments.